
DISPOSICION 556/2009
ADMINISTRACION NACIONAL DE MEDICAMENTOS, ALIMENTOS Y TECNOLOGIA MEDICA (A.N.M.A.T.)
| |
Apruébase la Guía para aplicar en los Cambios de Escala y Cambios Posteriores al Registro de Medicamentos Sujetos a Demostración de Bioequivalencia.
Del: 05/02/2009; Boletín Oficial: 26/02/2009
|
VISTO la Ley Nº 16.643, sus Decretos Reglamentarios Nros . 9763/64, 150/92 y 177/93, el Decreto Nº 1490/92, la Resolución ex Secretaría de Políticas, Regulación y Relaciones Sanitarias Nº 46/03, las Disposiciones ANMAT nros. 5330/97, 3185/99, 229/00, 3311/01, 2807/02, 4290/02, 5318/02, 7062/02 y 2446/07; y el Expediente Nº 1-47-1110-676-08-7 del Registro de esta Administración Nacional; y
CONSIDERANDO;
Que las precitadas normas, y las disposiciones complementarias dictadas en su consecuencia, constituyen el ordenamiento legal aplicable a la aprobación, registro y autorización de venta de las especialidades medicinales cuya elaboración, importación y comercialización en el país se desarrolla al amparo de los preceptos generales establecidos por la Ley Nº 16.463. Que la aplicación de las normas aludidas tiene como finalidad última la protección de la salud de la población mediante la adopción de un modelo fiscalizador de gestión que, sin perjuicio de la lectura objetiva de la información calificada, destine los mayores esfuerzos a la verificación continua de la eficacia, seguridad y calidad de los productos que aquella consume.
Que por Disposición ANMAT Nº 3185/99 se aprobaron las recomendaciones técnicas para la realización de estudios de equivalencia contenidas en el documento denominado: “Cronograma para exigencia de estudios de equivalencia entre medicamentos de riesgo sanitario significativo”.
Que es necesario establecer requisitos para la solicitud y posterior aprobación de los estudios de Biodisponibilidad/Bioequivalencia in vivo/in vitro, según corresponda, en los casos de cambios específicos respecto de: la composición del producto, el lugar de manufactura, la escala de manufactura (aumento o disminución) y/o en la elaboración (proceso y equipos), de una formulación oral de liberación inmediata.
Que todo cambio que soliciten los titulares de registro debe ser documentado y debidamente autorizado.
Que es necesario determinar los parámetros que permitan asegurar la continuidad de la calidad y del comportamiento de los productos medicinales en una forma farmacéutica oral sólida de liberación inmediata, para aquellos cambios antes señalados, que sean posteriores a su autorización en medicamentos comprendidos en las Disposiciones Nros. 3185/99, 3311/01, 2807/02, 229/00, 2446/07 y en todas aquellas que se establezca la obligatoriedad de demostración de Bioequivalencia.
Que a tales fines es necesario adoptar una Guía para aplicar a los Cambios de Escala y Cambios Posteriores al Registro de aquellos medicamentos, cuyos principios activos estén sujetos a la demostración de bioequivalencia, en las formas farmacéuticas de sólidos orales de liberación inmediata.
Que el procedimiento de declaración juratoria para solicitar la aprobación de un cambio de excipiente, previsto en la Disposición (ex Subsecretaría de Regulación y Control) Nº 853/89, resulta insuficiente para autorizar los cambios específicos señalados para los casos de formulaciones sólidas orales de liberación inmediata, comprendidas en el párrafo anterior.
Que tales solicitudes quedarán sujetas a la presente Disposición.
Que el Instituto Nacional de Medicamentos y la Dirección de Asuntos Jurídicos han tomado intervención en el ámbito de sus competencias.
Por ello;
EL INTERVENTOR
DE LA ADMINISTRACION NACIONAL DE MEDICAMENTOS ALIMENTOS
Y TECNOLOGIA MEDICA
DISPONE:
|
Artículo 1º - Apruébase la Guía para aplicar en los Cambios de Escala y Cambios Posteriores a las condiciones de autorización en el Registro de Especialidades Medicinales de aquellos productos cuyos principios activos están sujetos a demostración de Bioequivalencia en las formas farmacéuticas de Sólidos Orales de Liberación Inmediata, que figura como Anexo I de la presente Disposición, y forma parte integrante de la misma.
Art. 2º - Regístrese, comuníquese a quienes corresponda. Dése a la Dirección Nacional del Registro Oficial para su publicación; notifíquese a las entidades y organizaciones profesionales correspondientes. Cumplido, archívese PERMANENTE
ANEXO I
Guía para aplicar a
CAMBIOS DE ESCALA Y CAMBIOS POSTERIORES AL
REGISTRO DE MEDICAMENTOS SUJETOS A
DEMOSTRACION DE BIOEQUIVALENCIA.
Formas Farmacéuticas Sólidas Orales de Liberación Inmediata
Indice
I. PROPOSITO DE LA GUIA
II. GLOSARIO
III. CAMBIOS EN LA COMPOSICION
IV. CAMBIOS DE SITIO DE ELABORACION
V. CAMBIOS DE TAMAÑO DE LOTE (AUMENTO O DISMINUCION DE ESCALA)
VI. CAMBIOS EN LA ELABORACION
VII. REQUISITOS PARA CAMBIOS MULTIPLES
VIII. DISOLUCION IN VITRO
IX. ESTUDIOS DE BIOEQUIVALENCIA.
X. SISTEMA DE CLASIFICACION BIOFARMACEUTICA
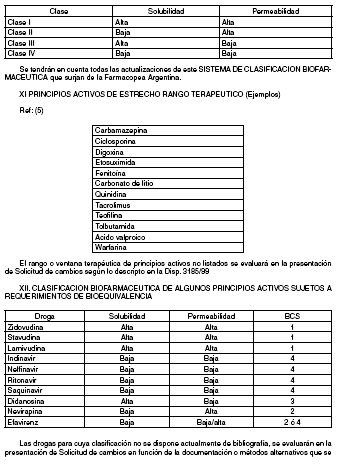
XI. PRINCIPIOS ACTIVOS DE ESTRECHO RANGO TERAPEUTICO (Ejemplos)
XII. CLASIFICACION BIOFARMACEUTICA DE ALGUNOS PRINCIPIOS ACTIVOS SUJETOS A
REQUERIMIENTOS DE BIOEQUIVALENCIA
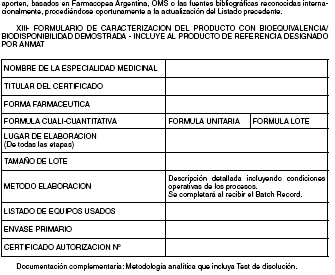
XIII. FORMULARIO DE CARACTERIZACION DE PRODUCTO CON BIOEQUIVALENCIA/BIODISPONIBILIDAD
DEMOSTRADA (INCLUYE AL PRODUCTO DE REFERENCIA DESIGNADO POR
ANMAT)
XIV. REFERENCIASCAMBIOS DE ESCALA Y CAMBIOS POSTERIORES AL REGISTRO DE
MEDICAMENTOS SUJETOS A DEMOSTRACION DE BIOEQUIVALENCIA
Formas Farmacéuticas Sólidas Orales de Liberación Inmediata
I. PROPOSITO DE LA GUIA
Esta guía establece los criterios y detalla la información requerida para las solicitudes de cambio
posteriores a la aprobación del estudio de biodisponibilidad/bioequivalencia in vivo/in vitro, según
corresponda, en cuanto a:
1) Cambio en la composición;
2) Cambio del lugar de manufactura;
3) Cambio en la escala de manufactura (aumento o disminución) y/o
4) Cambio en la elaboración (proceso y equipos) de una formulación oral de liberación inmediata.
Las únicas condiciones de elaboración y composición autorizadas por la Autoridad Sanitaria
serán las consignadas en el Formulario de Caracterización de Producto (Anexo XIII) En el caso del
producto test, corresponderán a las del biolote. Para los productos de referencia, corresponderán a
las vigentes al momento de su designación por ANMAT
Cualquier cambio debe ser documentado y debidamente autorizado.
Se definen:
1) los niveles de cambio;
2) los ensayos químicos recomendados para cada nivel de cambio;
3) los ensayos de disolución in vitro y/o los ensayos de bioequivalencia in vivo para cada nivel
de cambio y
4) la documentación que debería respaldar el cambio solicitado.
Este documento enuncia la información que debería proporcionarse a la ANMAT en la solicitud,
para asegurar la continuidad de la calidad y comportamiento del producto en una forma
farmacéutica oral sólida de liberación inmediata, para los cambios especificados posteriores a la
aprobación.
No afecta ningún cambio posterior a la aprobación fuera de los especificados. Para los cambios
no tratados en esta guía, los solicitantes deberán contactar a la ANMAT/INAME para obtener información
acerca de la factibilidad y documentación necesaria.
Para cambios posteriores a la aprobación de la bioequivalencia de formas farmacéuticas de liberación
inmediata que afectan a la composición, cambios de escala, cambios de sitio de elaboración
y cambios en los procesos o equipos de manufactura esta guía reemplaza a la Disposición (SRyC)
Nº 853/89 únicamente en aquellos medicamentos comprendidos en las Disposiciones Nros. 3185/99,
3311/01, 2807/02, 229/00, 2446/2007 y en toda aquella que establezca obligatoriedad de demostración
de bioequivalencia.
Se incluyen en el alcance de esta Guía a los Productos de referencia establecidos por ANMAT
que sufrieran cambios posteriores a la emisión de la respectiva Disposición.
Los productos de referencia establecidos actualmente por ANMAT deberán aportar la información
que cumplimente el formulario del Anexo XIII.
II. GLOSARIO
Biolote: Lote con el cual se realizó el estudio de biodisponibilidad/bioequivalencia aprobado.
Calidad técnica de un excipiente: Conjunto de atributos y/o especificaciones evaluados a través
de procedimientos de caracterización química, física (por ejemplo granulometría, hábito cristalino,
etc.) y microbiológica. Puede abarcar parámetros con posible impacto en la calidad y comportamiento
de la forma farmacéutica, que no sean habitualmente determinados en el control de calidad de la
materia prima en cuestión.
Cambio de calidad técnica: Cualquier cambio en alguno de los atributos y/o especificaciones que
la definen. Quedan expresamente excluidos los cambios de un procedimiento analítico que sea equivalente
o superior al modificado sin ampliación de los límites de aceptabilidad, así como los cambios
derivados de la actualización de una monografía de Farmacopea.
Equipos: Utensilios, instrumentos y aparatos, automatizados o no automatizados, mecánicos o
no mecánicos, utilizados para elaborar el medicamento, incluyendo los utilizados para su envasado.
Escala:
Aumento de escala: Proceso de aumentar el tamaño del lote.
Disminución de escala: Proceso de disminuir el tamaño del lote.
Escala piloto: La elaboración de un medicamento, por medio de un procedimiento plenamente
representativo, que simula el utilizado para la escala industrial.
Para formas farmacéuticas orales sólidas, el tamaño del lote piloto deberá ser de, como mínimo,
la décima parte de la escala industrial, o 10.000 comprimidos o cápsulas, el número que resulte
mayor.
Estabilidad acelerada: Cuando la guía solicita estabilidad acelerada, deberán presentarse datos
de un estudio de tres meses realizado sobre la formulación aprobada, y de 1 lote con 3 meses de
estabilidad acelerada, de la formulación con el cambio que se está solicitando. Las condiciones del
estudio deberán ser: 40ºC ± 2ºC y 75% RH ± 5% RH.
La frecuencia de análisis será de 0, 1, 2 y 3 meses. La evaluación de los datos se realizará en
base al porcentaje de caída del principio activo y/o al aumento de sustancias relacionadas.
Excipientes multifuncionales: Son aquellos que cumplen funciones múltiples en una determinada
formulación. Por ejemplo: Almidón como diluyente y desintegrante.
Formulación: Listado de los ingredientes y composición de la forma farmacéutica. medicinales
que posean más de tres años de experiencia comercial del producto registrado.
Lote: Cantidad definida de materia prima, material de acondicionamiento o producto, elaborado
en un proceso o serie de procesos de forma tal que sea homogénea.
A fines de control del producto terminado, un lote de un producto farmacéutico comprende todas
las unidades de una forma farmacéutica producidas a partir de la misma masa inicial de materiales, y
que ha sufrido una única serie de operaciones de fabricación o una sola operación de esterilización o,
en caso de proceso de producción continua, todas las unidades fabricadas en un período de tiempo
determinado. Con el fin de realizar ciertas fases de la elaboración, puede ser necesario dividir un lote
en diversos sub-lotes, que se unen después para constituir un lote final homogéneo. En caso de elaboración
continua, el lote debe corresponder a una fracción definida de la producción, caracterizada
por su homogeneidad prevista.
Mecanismo operativo: Reglas o conceptos que gobiernan la operación del sistema.
Medicamento: Toda preparación o producto farmacéutico empleado para la prevención, diagnóstico
y/o tratamiento de una enfermedad o estado patológico, o para modificar sistemas fisiológicos en
beneficio de la persona a quien se le administra. (F.A.VII)
Principio activo: Toda sustancia química o mezcla de sustancias relacionadas, de origen natural
o sintético, que poseyendo un efecto farmacológico específico, se emplea en medicina humana.
(F.A.VII)
Proceso: Conjunto de acciones seguidas para llegar al resultado esperado.
Rango: Medida, o límites entre los cuales las variaciones son aceptables.
Rango terapéutico estrecho o estrecha ventana terapéutica: Definición según Disposición (ANMAT)
Nº 3185/99.
Validación: Acción documentada, en concordancia con los principios de las Buenas Prácticas de
Fabricación, que demuestra que los procedimientos, procesos, equipamientos, materiales, actividades
o sistemas conducen realmente a los resultados previstos.
III. CAMBIOS EN LA COMPOSICION
Esta sección de la guía enfoca los cambios de excipientes del medicamento. Esta guía no considera
cambios referidos al principio activo. Los cambios en la composición que tienen el efecto de
agregar o eliminar un excipiente, se definen en el Nivel 3. Para excipientes multifuncionales1[1] se le
asignará la categoría funcional con menor porcentaje de cambio permitido. (Ej. Almidón como diluyente
y desintegrante, tendrá un porcentaje de cambio máximo permitido del 3% en Nivel 1).
A. Cambios de Nivel 1
1. Definición de Nivel: Los cambios de Nivel 1 son aquellos con poca probabilidad de tener un
impacto detectable en la calidad y comportamiento de la formulación.
Ejemplos:
a. Eliminación total o parcial de un ingrediente, cuyo propósito es afectar el color o el sabor del
medicamento; o un cambio en el ingrediente de la tinta de impresión por otro ingrediente aprobado.
b. Cambios en excipientes, expresados como un porcentaje (p/p) de la formulación total, menores
o iguales a los siguientes rangos:
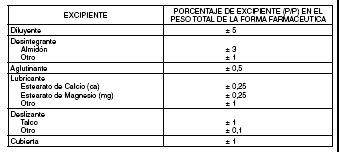
Estos porcentajes se basan en el supuesto de que la droga en el producto está formulada al
100% de lo declarado o de la potencia. El efecto aditivo total de todos los cambios de excipientes no
deberá superar el 5%. Ejemplo:
En un producto que consiste en el ingrediente activo A, lactosa, celulosa microcristalina y estearato
de magnesio; la lactosa y la celulosa microcristalina no deberán variar más de un 5% del total
absoluto (por ejemplo, la lactosa aumenta un 2,5% y la celulosa microcristalina disminuye un 2,5%)
en relación con el peso de la forma farmacéutica, si ha de quedar dentro del rango del Nivel 1.
Para establecer los porcentajes del cambio de excipientes propuesto se tomará como referencia
la composición cuali-cuantitativa aprobada del lote que fue utilizado para la demostración de bioequivalencia/
y no la resultante de un cambio.
2. Documentación a ser presentada en la solicitud de cambio.
a. Ficha de modificaciones.
b. Declaración de la función que cumple en la formulación cada uno de los excipientes.
c. Porcentaje de cambio de excipientes, individual y total, respecto del peso total de la forma
farmacéutica, que justifiquen la inclusión en Nivel 1.
d. Documentación Química: Especificaciones aprobadas para producto terminado.
e. Documentación de Disolución: Especificaciones aprobadas para producto terminado.
f. Registro Maestro. g. Documentación de Bioequivalencia: No se requiere.
3- Documentación disponible en el Laboratorio para la Autoridad Sanitaria.
a. Documentación de estabilidad: Estudio de estabilidad en curso de un lote industrial elaborado
con la nueva formulación propuesta, con seguimiento por el lapso de vida útil.
B. Cambios de Nivel 2
1. Definición de Nivel: Los cambios de Nivel 2 son aquellos que podrían tener un impacto significativo
en la calidad y el comportamiento de la formulación.
Los ensayos y la documentación para la presentación de un cambio de Nivel 2, varían según tres
factores: rango terapéutico, solubilidad y permeabilidad.
El capítulo XI contiene a modo de ejemplo un listado de principios activos de estrecho rango terapéutico
incluidos en las Disposiciones que requieren bioequivalencia. La solubilidad y la permeabilidad
se definen como “alta” o “baja” según el Capítulo X.
Ejemplos:
a. Cambio en la calidad técnica de un excipiente. Ejemplo Celulosa microcristalina PH102 vs.
Celulosa microcristalina PH 200.
b. Cambios en los excipientes, expresados como porcentajes (p/p) de la formulación total, mayores
que los que figuran anteriormente para un cambio de Nivel 1, pero menores o iguales a los
siguientes rangos porcentuales (que representan un aumento dos veces mayor que los cambios de
Nivel 1).
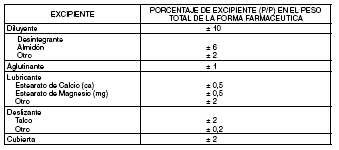
Estos porcentajes se basan en el supuesto de que el principio activo en el producto está formulado
al 100% de lo declarado o de la potencia. El efecto aditivo total de todos los cambios de excipientes
no deberá superar el 10%.
Para establecer los porcentajes del cambio de excipientes propuesto se tomará como referencia
la composición cuali-cuantitativa aprobada del lote que fue utilizado para la demostración de
bioequivalencia y no la resultante de cambios anteriores de composición ya sea de Nivel 1 o
Nivel 2.
2. Documentación a ser presentada en la solicitud de cambio.
a. Ficha de modificaciones
b. Declaración de la función que cumple en la formulación cada uno de los excipientes.
c. Porcentaje de cambio de excipientes, individual y total, respecto del peso total de la forma
farmacéutica, que justifiquen la inclusión en Nivel 2.
d. Documentación Química: Especificaciones aprobadas para producto terminado
e Documentación de Disolución
e 1 Drogas de alta permeabilidad y alta solubilidad. Clase I
Caso A: Según Capítulo VIII. DISOLUCION IN VITRO
Disolución del 85% a los 15 minutos, en 900 mL de HCl 0,1N.
Si un medicamento no cumple con este criterio, el solicitante deberá realizar los ensayos descriptos
para el Caso B y de no cumplirlos, los del Caso C.
e.2 Drogas de baja permeabilidad y alta solubilidad. Clase III
Caso B: Según Capítulo VIII. DISOLUCION IN VITRO Se deberá realizar un perfil de disolución
de puntos múltiples en el medio aprobado o codificado, a los 15, 30, 45, 60 y 120 minutos, o hasta que
se logre una asíntota. Los perfiles de disolución de las formulaciones actuales y de las propuestas
deberán ser similares.
e.3 Drogas de alta permeabilidad y baja solubilidad. Clase II
Caso C: Según Capítulo VIII. DISOLUCION IN VITRO: Perfiles múltiples. Se deberá realizar un
muestreo apropiado a los 15, 30, 45, 60 y 120 minutos, hasta que se disuelva el 90% de la droga o
hasta que se logre una asíntota. Se puede utilizar un tensioactivo siempre que su uso se justifique
apropiadamente.
Los perfiles de disolución de las formulaciones actuales y propuestas, deberán ser similares.
f. Registro Maestro.
g. Documentación de estabilidad: Estudio de estabilidad de un lote con tres meses de datos de
estabilidad acelerada de la formulación aprobada y 1 lote con 3 meses de estabilidad acelerada, de
la formulación con el cambio solicitado.
h. Documentación de Bioequivalencia: Ninguna. De no cumplirse con lo requerido como caso A,
B o C de Disolución, deberá cumplimentarse lo solicitado en Bioequivalencia para Nivel 3. 3- Documentación disponible en el laboratorio para la Autoridad Sanitaria
a. Documentación de estabilidad: Estudio de estabilidad en curso de un lote industrial elaborado
con la nueva formulación propuesta, con seguimiento por el lapso de vida útil.
C. Cambios de Nivel 3
1. Definición de Nivel: Los cambios de Nivel 3 son aquellos con alta probabilidad de tener un
impacto significativo en la calidad y comportamiento de la formulación. La documentación a presentar
varía según los siguientes factores: rango terapéutico solubilidad y permeabilidad.
Ejemplos:
a. Todo cambio cualitativo y cuantitativo en los excipientes que acompañan a una droga de estrecho
rango terapéutico, más allá de lo señalado en la Sección III.A.1.b.
b. Cualquier producto que no cumpla las exigencias de disolución bajo la Sección III.B.2.e
c. Cambios en excipientes de drogas de baja solubilidad y baja permeabilidad, más allá de los
rangos señalados en la Sección III.A.1.b.
d. Cambios en excipientes de todas las drogas, más allá de los rangos señalados en la Sección
III.B.1.b.
2. Documentación a ser presentada en la solicitud de cambio.
a. Ficha de modificaciones.
b. Declaración de la función que cumple en la formulación cada uno de los excipientes.
c. Porcentaje de cambio de excipientes, individual y total, respecto del peso total de la forma
farmacéutica.
d. Registro Maestro.
e. Documentación de Disolución.
Caso B: Se deberá realizar un perfil de disolución de puntos múltiples en el medio aprobado o
codificado, a los 15, 30, 45, 60 y 120 minutos, o hasta que se logre una asíntota. Los perfiles de disolución
de las formulaciones actuales y de las propuestas deberán ser similares.
f. Documentación Química: Especificaciones aprobadas para producto terminado.
g. Documentación de Bioequivalencia: Estudio completo de bioequivalencia in vivo o in vitro
según corresponda
El estudio de bioequivalencia podrá ser obviado cuando exista una correlación in vivo/in Vitro
aceptable, que haya sido verificada.
h. Documentación de estabilidad:
h.1. Información relevante disponible: Un lote con tres meses de datos de estabilidad acelerada
de la formulación aprobada y 1 lote con 3 meses de estabilidad acelerada, de la formulación con el
cambio solicitado.
h.2 Información relevante no disponible: Tres lotes con tres meses de datos de estabilidad acelerada
de la formulación aprobada y 3 lotes con 3 meses de estabilidad acelerada, de la formulación
con el cambio solicitado.
3- Documentación disponible en el Laboratorio para la Autoridad Sanitaria.
a. Documentación de estabilidad: Estudio de estabilidad en curso de un lote industrial con la
nueva formulación, por el lapso de vida útil.
IV. CAMBIOS DE SITIO DE ELABORACION
Los cambios de sitio de elaboración, consisten en cambios en la ubicación del lugar de manufactura
para instalaciones, tanto de propiedad de la empresa, como contratadas, y no incluyen ningún
cambio de escala, cambio de manufactura (incluyendo el proceso y/o los equipos) ni cambios en la
composición. La Sección V de esta guía trata los aumentos de escala. Los nuevos sitios de elaboración
deberán tener una inspección de Buenas Prácticas de Manufactura (GMP) satisfactoria.
A. Cambios de Nivel 1
1. Definición de Nivel: Los cambios de Nivel 1 consisten en cambios de sitio de elaboración
dentro de un establecimiento único, donde se utilizan los mismos equipos, procedimientos operativos
estándar (POEs), condiciones ambientales (ejemplo: temperatura y humedad) y controles, así como
personal común a ambos sitios de elaboración o que posean experiencia adecuada con el proceso
de elaboración, y donde no se realiza ningún cambio en los batch records, excepto la información
administrativa y la ubicación del establecimiento.
2. Documentación a ser presentada en la solicitud de cambio
a. Ficha de modificaciones y aclaración sobre la naturaleza del cambio propuesto.
b. Documentación Química: Especificaciones aprobadas para producto terminado.
c. Documentación de Disolución: Especificaciones aprobadas para producto terminado.
d. Documentación de Bioequivalencia: No se requiere
B. Cambios de Nivel 2
1. Definición de Nivel: Los cambios de Nivel 2 consisten en traslados o mudanzas a otro establecimiento,
donde se utilizan los mismos equipos, POEs, condiciones ambientales (ejemplo: temperatura
y humedad) y controles, así como personal común a ambos sitios de manufactura, y donde no
se realiza ningún cambio en los batch records, excepto información administrativa y la ubicación del
establecimiento.
2. Documentación a ser presentada en la solicitud de cambio.
a. Ficha de modificaciones. Ubicación del nuevo sitio. b. Documentación Química: Especificaciones aprobadas para producto terminado.
c. Registro Maestro.
d. Documentación de Disolución: Especificaciones aprobadas para producto terminado.
e. Documentación de Bioequivalencia: No se requiere.
3. Documentación disponible en el Laboratorio para la Autoridad Sanitaria
a. Documentación de estabilidad: Estudio de estabilidad en curso de un lote industrial elaborado
en el nuevo sitio, con seguimiento por el lapso de vida útil.
C. Cambios de Nivel 3
1. Definición de Nivel: Los cambios de Nivel 3 consisten en traslados o mudanzas a otro establecimiento,
donde se utilizan diferentes equipos y personal.
La modificación en los equipos y procesos no deberá ser mayor a la establecida en el Nivel 1 de.
VI. Elaboración.
2. Documentación a ser presentada en la solicitud de cambio
a. Ficha de modificaciones.
b. Habilitación del nuevo sitio de elaboración.
c. Registro Maestro actualizado.
d. Informe detallado del cambio de equipos y/o procesos.
e. Documentación de Disolución:
Caso B Se deberá realizar un perfil de disolución de puntos múltiples en el medio aprobado o
codificado, a los 15, 30, 45, 60 y 120 minutos, o hasta que se logre una asíntota. Los perfiles de disolución
en las condiciones actuales y las propuestas deberán ser similares.
f. Documentación Química: Especificaciones aprobadas para producto terminado.
g. Documentación de estabilidad:
g.1 Información relevante disponible: Un lote con tres meses de datos de estabilidad acelerada
de la formulación aprobada y 1 lote con 3 meses de estabilidad acelerada, de la formulación con el
cambio solicitado.
g.2 Información relevante no disponible: Tres lotes con tres meses de datos de estabilidad acelerada
de la formulación aprobada y 3 lotes con 3 meses de estabilidad acelerada, de la formulación
con el cambio solicitado.
h- Documentación de Bioequivalencia: No se requiere.
3- Documentación disponible en el Laboratorio para la Autoridad Sanitaria.
a. Documentación de estabilidad: Estudio de estabilidad en curso de un lote industrial elaborado
en el nuevo sitio, con seguimiento por el lapso de vida útil.
V. CAMBIOS EN EL TAMAÑO DEL LOTE (CAMBIOS DE ESCALA)
Esta sección de la guía enfoca los cambios referentes al tamaño de un lote, ya sea aumento
o disminución del mismo, respecto al lote utilizado en la demostración de bioequivalencia/equivalencia
in vitro (según corresponda). La disminución de escala por debajo de las 10.000 unidades
posológicas no está contemplada en esta guía. Todos los aumentos de escala deberán ser debidamente
validados y, en caso de ser necesario, inspeccionados por personal de la Autoridad
Sanitaria.
A. Cambios de Nivel 1
1. Definición de Nivel: Cambio en tamaño de lote, hasta un factor de 10 veces el tamaño del
lote utilizado en la demostración de bioequivalencia/equivalencia in vitro (según corresponda),
donde:
a. Los equipos utilizados tienen el mismo diseño y principios operativos.
b. Los lotes se elaboran en pleno cumplimiento de las GMP vigentes, y
c. Se utilizan los mismos POEs y controles, así como la misma formulación y procedimientos de
elaboración.
2. Documentación a ser presentada en la solicitud de cambio.
a. Ficha de modificaciones detallando el cambio de escala.
b. Registro Maestro.
c. Documentación Química: Especificaciones aprobadas para producto terminado.
d. Documentación de Disolución: Especificaciones aprobadas para producto terminado.
e. Documentación de Bioequivalencia: No se requiere..
3- Documentación disponible en el Laboratorio para la Autoridad Sanitaria:
a. Documentación de estabilidad: Estudio de estabilidad en curso de un lote industrial con el
nuevo tamaño, con seguimiento por el lapso de vida útil.
B. Cambios de Nivel 2
1. Definición de Nivel: Cambio en tamaño de lote, más allá de un factor de 10 veces el tamaño
del lote utilizado en la demostración de bioequivalencia, donde:
a. Los equipos utilizados tienen el mismo diseño y principios operativos;
b. los lotes se elaboran en pleno cumplimiento de las GMP vigentes, yc. se utilizan los mismos POEs y controles, así como la misma formulación y procedimientos de
elaboración utilizados en el lote empleado en la demostración de bioequivalencia.
2. Documentación a ser presentada en la solicitud de cambio.
a. Ficha de modificaciones detallando el cambio de escala.
b. Registro Maestro actualizado.
c. Documentación de Disolución:
Caso B: Se deberá realizar un perfil de disolución de puntos múltiples en el medio aprobado
o codificado, a los 15, 30, 45, 60 y 120 minutos, o hasta que se logre una asíntota. Los perfiles de
disolución de los lotes de diferente tamaño deberán ser similares.
d. Documentación Química: Especificaciones aprobadas para producto terminado.
e. Documentación de estabilidad: Estudio de estabilidad de un lote con tres meses de datos de
estabilidad acelerada de la formulación aprobada y 1 lote con 3 meses de estabilidad acelerada, de
la formulación con el cambio solicitado.
f Documentación de Bioequivalencia: No se requiere.
3- Documentación disponible en el Laboratorio para la Autoridad Sanitaria.
a. Documentación de estabilidad: Estudio de estabilidad en curso de un lote industrial elaborado
con el nuevo tamaño, con seguimiento por el lapso de vida útil.
VI. CAMBIOS EN LA ELABORACION
Los cambios de elaboración podrán afectar tanto a los equipos utilizados en el proceso de manufactura
como al proceso en sí.
A. Cambios de Nivel 1
1. Defunción de Nivel: Esta categoría comprende:
a. Cambios de proceso de elaboración, ya sea en tiempos de mezcla o de velocidades de operación,
y/o
b. Cambios a equipos alternativos, con el mismo diseño y principios operativos, y con igual o
distinta capacidad.
2. Documentación a ser presentada en la solicitud de cambio:
a. Ficha de modificaciones detallando el cambio de proceso y/o equipo
b. Registro Maestro.
c. Documentación Química: Especificaciones aprobadas para producto terminado.
d. Documentación de Disolución: Especificaciones aprobadas para producto terminado.
e. Documentación de Bioequivalencia: Ninguna.
3- Documentación disponible en el Laboratorio para la Autoridad Sanitaria:
a. Documentación de estabilidad: Estudio de estabilidad en curso de un lote industrial elaborado
con los cambios propuestos, con seguimiento por el lapso de vida útil.
B. Cambios de Nivel 2
1. Definición de Nivel: Esta categoría comprende:
a. Cambios diferentes a tiempos de mezcla o de velocidades de operación en el proceso de
elaboración, que no impliquen un cambio en el tipo de proceso, y/o
b. cambio en el diseño y/o en los principios operativos de los equipos utilizados.
Los ensayos y la documentación para la presentación de un cambio de Nivel 2, varían según tres
factores: rango terapéutico, solubilidad y permeabilidad.
2. Documentación a ser presentada en la solicitud de cambio.
a. Ficha de modificaciones con la información detallada del cambio propuesto.
b. Registro Maestro actualizado.
c. Documentación de estabilidad: Estudio de estabilidad de un lote con tres meses de datos de
estabilidad acelerada de la formulación aprobada y 1 lote con 3 meses de estabilidad acelerada, de
la formulación con el cambio solicitado.
d. Documentación de Disolución:
d.1 Drogas de alta permeabilidad y alta solubilidad.
Caso A: Disolución del 85% a los 15 minutos, en 900 mL de HCl 0,1N.
Si un medicamento no cumple con este criterio, el solicitante deberá realizar los ensayos descriptos
para el Caso B o C.
d.2 Drogas de baja permeabilidad y alta solubilidad.
Caso B: Se deberá realizar un perfil de disolución de puntos múltiples en el medio aprobado o
codificado, a los 15, 30, 45, 60 y 120 minutos, o hasta que se logre una asíntota. Los perfiles de disolución
de las formulaciones actuales y de las propuestas deberán ser similares.
d.3 Drogas de alta permeabilidad y baja solubilidad.
Caso C: Se deberá realizar un muestreo apropiado a los 15, 30, 45, 60 y 120 minutos, hasta que
se disuelva el 90% de la droga o hasta que se logre una asíntota. Se puede utilizar un tensioactivo
siempre que su uso se justifique apropiadamente. Los perfiles de disolución de las formulaciones
actuales y propuestas, deberán ser similares. e. Documentación Química: Especificaciones aprobadas para producto terminado.
f. Documentación de Bioequivalencia: Ninguna. De no cumplirse con lo requerido como caso A, B
o C de Disolución, deberá cumplimentarse lo solicitado en Bioequivalencia para Nivel 3.
3- Documentación disponible en el Laboratorio para la Autoridad Sanitaria:
a. Documentación de estabilidad: Estudio de estabilidad en curso de un lote industrial elaborado
con los cambios propuestos, con seguimiento por el lapso de vida útil.
C. Cambios de Nivel 3
1. Definición de Nivel: Esta categoría comprende:
a. Cambios en el tipo de proceso utilizado en la elaboración del producto (ejemplo: granulado por
vía húmeda vs. compresión directa).
b. Cualquier producto que no cumpla las exigencias de disolución bajo la Sección V.B.2
c. Modificaciones en la elaboración de productos con principios activos de baja solubilidad y baja
permeabilidad, más allá de lo admitido en Nivel 1 (VI. A)
d. Modificaciones en la elaboración de productos con principios activos de rango terapéutico
estrecho, más allá de lo admitido en Nivel 1 (VI. A)
2. Documentación a ser presentada en la solicitud de cambio:
a. Ficha de modificaciones detallando el cambio de proceso y/o equipo.
b. Registro Maestro actualizado.
c. Documentación Química: Especificaciones aprobadas para producto terminado.
d. Documentación de estabilidad:
d.1 Información relevante disponible: Un lote con tres meses de datos de estabilidad acelerada
de la formulación aprobada y 1 lote con 3 meses de estabilidad acelerada, de la formulación con el
cambio solicitado.
d.2 Información relevante no disponible: Tres lotes con tres meses de datos de estabilidad acelerada
de la formulación aprobada y 3 lotes con 3 meses de estabilidad acelerada, de la formulación
con el cambio solicitado.
e. Documentación de Disolución.
Caso B: Se deberá realizar un perfil de disolución de puntos múltiples en el medio aprobado o
codificado, a los 15, 30, 45, 60 y 120 minutos, o hasta que se logre una asíntota. Los perfiles de disolución
de las formulaciones actuales y de las propuestas deberán ser similares.
f- Documentación de Bioequivalencia: Estudio completo de bioequivalencia in vivo o in vitro según
corresponda. El estudio de bioequivalencia podrá ser obviado cuando exista una correlación in
vivo /in vitro aceptable, que haya sido verificada.
3- Documentación disponible en el Laboratorio para la Autoridad Sanitaria
a. Documentación de estabilidad: Estudio de estabilidad en curso de un lote industrial elaborado
con los cambios propuestos, con seguimiento por el lapso de vida útil.
VII. REQUISITOS PARA CAMBIOS MULTIPLES
Se entiende por cambios múltiples a las modificaciones en la escala y/o cambios posteriores al
registro de Especialidades Medicinales sujetas a demostración de bioequivalencia, presentados en
forma sucesiva o simultánea.
El Nivel correspondiente al cambio de excipiente y/o de tamaño de lote presentado, se determinará
en forma acumulativa tomando como referencia la información consignada en el Formulario de
Caracterización de Producto.
La documentación y ensayos exigibles para un cambio múltiple serán los correspondientes al de
mayor exigencia entre los cambios individuales que lo componen.
VIII. DISOLUCION IN VITRO
Esta Guía recomienda la comparación de perfiles de disolución, realizados en condiciones idénticas,
utilizando el Factor de Similitud f2 para aprobar los diversos niveles de cambio y documentar la
similitud entre el producto de prueba posterior al cambio y el de referencia anterior al cambia
donde R t y T t representan el porcentaje disuelto a cada tiempo y n es el número de puntos de
toma de muestra durante el ensayo de disolución. (n debe ser al menos igual a 3 y no debe incluir el
tiempo cero).
Procedimiento para determinar f2
1 - Determinar el perfil de disolución de los productos de prueba (posterior al cambio) y referencia
(anterior al cambio) utilizando 12 unidades de cada uno.
2 - Usando los valores de disolución medios de ambas curvas en cada intervalo temporal calcular
el factor de similitud f2.
3 - Un valor f2 comprendido entre 50 y 100, indica que los dos perfiles de disolución son similares.
Las mediciones de disolución tanto para el producto de prueba como para el de referencia deberán
realizarse bajo exactamente las mismas condiciones. Los tiempos de muestreo deberán ser
los mismos. Sólo se deberá considerar una medición después de la disolución del 85% de cada uno de ellos.
El coeficiente porcentual de variación en los puntos de toma de muestra más tempranos (Por ej. 15
min) no deberá ser mayor al 20%, y en los otros puntos no deberá ser mayor al 10%.
Ensayos de disolución
Caso A: Disolución de Q ≥ 85% a los 15 minutos en 900 mL de HCl 0,1N usando el Aparato 1 de
la USP <711> a 100 rpm, o el Aparato 2 a 50 rpm.
Caso B: Perfil de disolución de puntos múltiples realizados únicamente con el método aprobado
en el Registro o el codificado (*), a intervalos regulares hasta que se logre una asíntota para la formulación
propuesta y la aprobada. Aplicar Factor de Similitud f2.
Caso C: Perfiles de disolución de puntos múltiples realizados en HCL 0,1N y en buffers USP de
pH 4,5 y 6,8 tres perfiles distintos, para las formulaciones propuestas y actualmente aceptadas. Se
deberá realizar un muestreo apropiado, por ejemplo a los 15, 30, 45, 60 y 120 minutos, hasta que
se disuelva el 90% de la droga o hasta que se logre una asíntota. Se puede utilizar un tensioactivo
siempre que su uso sea justificado apropiadamente.
(*) Si la prueba de disolución ha sido incluida en ediciones de FA o USP posteriores al Registro,
se evaluará individualmente la metodología empleada.
IX. ESTUDIOS DE BIOEQUIVALENCIA
Los estudios de bioequivalencia in vivo/in vitro según corresponda, se realizarán de acuerdo a los
procedimientos descriptos en la Disposición ANMAT Nº 3185/99 o en la que su defecto la reemplace
y/o complemente, utilizando como comparador el producto elaborado antes del cambio propuesto.
X. SISTEMA DE CLASIFICACION BIOFARMACEUTICA
En formas farmacéuticas de administración oral, los parámetros fundamentales que controlan
la velocidad y cantidad de principio activo absorbido, son la solubilidad acuosa y la permeabilidad
gastrointestinal. El sistema de clasificación biofarmacéutica (Biopharmaceutic drug clasification system
(BCS)) clasifica los principios activos en cuatro categorías de acuerdo a su solubilidad y permeabilidad.
Para determinar si una droga es de alta o baja solubilidad, se establece la relación entre
la concentración posológica mayor del fármaco y la menor solubilidad de la droga dentro del rango
fisiológico de pH 1,2 a 6,8 (Ref. 4) y en el de temperatura (37 ± 0,5º C). Las drogas de alta solubilidad
son aquellas con un volumen resultante de dividir la dosis máxima por la solubilidad mínima, que
sea menor o igual a 250 mL. Ejemplo: La solubilidad más baja del compuesto A (1,0 mg/raL), ocurre
a 37 ± 0,5º C y a pH 6,8; y se encuentra en el mercado en concentraciones de 100 mg, 200 mg y
400 mg. Esta droga se consideraría de baja solubilidad, ya que su volumen de dosis/solubilidad es
mayor de 250 mL (400 mg/1,0 mg/mL = 400 mL). La permeabilidad, Pe (cm/seg), se define como la
permeabilidad eficaz de la pared del yeyuno humano a una droga; e incluye una resistencia aparente
al transporte de masa hacia la membrana intestinal. Una droga se considera altamente permeable
cuando la fracción de absorción es igual o mayor al 85% de la dosis administrada, dato obtenido por
un estudio de balance de masas o en comparación a una administración intravenosa en humanos,
alternativamente otros métodos pueden ser utilizados.
XIV. REFERENCIAS
1. Scale-up and Post Approval Chages Guidance for Inmediate Release Products (SUPAC-IR)
US Food and Drug Administration-Centre for Drug Evaluation and Research 1995.
2. Classification of orally administered drugs on the World Health Organization Model list of Essential
Medicines according to the biopharmaceutics classification system. Lindenberg M. et al. Eur.
J.of Pharm. and Biopharm.58:265-278, 2004.
3. Molecular properties of WHO essential drugs and provisional biopharmaceutical classification.
Kasim N. et al. Mol. Pharmac. 1:85-95, 2003.
4. WHO Technical Report Series-40th Report-Anexo 8 Proposal to waive in vivo bioequivalence
requirements for WHO list of essential medicines inmediate release, solid oral dosage forms.
5. Japanese Regulation: Guideline for bioequivalence studies for formulation changes of oral dosage
forms. February 2000.
|

